I tumori endocrini del Pancreas
Le neoplasie che originano dal pancreas endocrino (Isole di Langerhans) sono rare con un’incidenza annuale approssimativa di cinque casi per milione d’abitanti per anno. Esse sono comunemente distinte in tumori funzionanti, se in grado di produrre e versare nel sangue particolari sostanze (ormoni) responsabili di una sindrome clinica (insieme di sintomi e segni particolari), e non funzionanti se non sono in grado di farlo. I tumori endocrini del pancreas possono essere benigni o maligni; questi ultimi a loro volta con bassa o alta aggressività. In ogni caso la sopravvivenza, a parità di stadio della malattia, è nettamente più lunga rispetto al classico carcinoma pancreatico. Per questo motivo, e perché il trattamento chirurgico o medico è completamente diverso, quando ci si trova di fronte ad una neoplasia pancreatica è consigliabile giungere alla conferma istologica (dopo asportazione chirurgica ogni qualvolta possibile o con biopsia percutanea nei casi non operabili) per evitare errori nella scelta del trattamento. I tumori endocrini sia benigni che maligni mostrano al microscopio caratteristiche istologiche simili, per cui, per determinare la malignità della lesione, è necessario documentare la presenza di un’invasione locale, e/o di metastasi ai linfonodi, al fegato o ad altri organi a distanza (polmoni, cervello, ossa). Il trattamento di un paziente con sospetto tumore endocrino del pancreas segue alcuni principi generali:
- Ricerca e caratterizzazione dell’eventuale produzione ormonale e, quando presente, della sindrome clinica. Si può avere una ipersecrezione ormonale in assenza di una sindrome clinica (sindrome allo stadio sub-clinico). Esiste una sindrome clinica tipica per l’insulinoma, il gastrinoma, il glucagonoma, il vipoma, il somatostinoma, l’ACTHoma, etc. Ci sono tuttavia molti tumori endocrini non associati a sindrome clinica. Essi sono abitualmente definiti “non funzionanti”. E’ possibile che tumori endocrini non funzionanti con il tempo manifestino una sindrome clinica.
- Dosaggio dei marcatori tumorali dei tumori endocrini: Cromogranina A [fare attenzione ai falsi positivi dovuti all’assunzione di farmaci (antiH2, inibitori di pompa protonica) o a gastrite atrofica], Enolasi Neuronale Specifica (NSE).
- Localizzazione, stadiazione accurata della neoplasia e valutazione della sua resecabilità. La localizzazione e la stadiazione dei tumori endocrini pancreatici sono eseguite con le tradizionali metodiche d’immagine: l’ecografia, la TAC spirale, la RMN e l’ecoendoscopia. L’accuratezza di tali metodiche varia in funzione della sede e delle dimensioni della lesione oltre che dell’esperienza dell’operatore; è possibile anche valutare l’eventuale interessamento dei linfonodi e la presenza di metastasi al fegato. A differenza del carcinoma esocrino, si può sfruttare la presenza di recettori cellulari per la somatostatina in molti tumori endocrini per eseguire un esame scintigrafico (Octreoscan) o PET (Tomografia a Emissione di Positroni, in questo caso si tratta della 68Ga-DOTANOC PET/TAC), utilizzando dei derivati della somatostatina marcati con apposite sostanze radioattive. In questo modo è possibile identificare sia la sede del tumore primitivo sia eventuali metastasi a distanza (fegato, linfonodi, polmoni, ossa, etc.). In caso di metastasi epatiche è utile una biopsia percutanea per caratterizzare l’aggressività del tumore (bassa, intermedia, alta), ma un aiuto può essere fornito dalla PET/TAC con 18 Fluorodesossiglucosio (18F-FDG PET/TAC), che consente di identificare i tumori a elevata attività metabolica e più aggressivi grazie al loro maggiore consumo di glucosio.
- Esclusione (o conferma) di una forma familiare (adenomatosi multi endocrina, MEN1: vedi più avanti) sulla base dell’analisi dell’albero genealogico e del dosaggio di una serie di ormoni prodotti da altre ghiandole endocrine.
Il trattamento dei tumori endocrini è costituito da quattro componenti: chirurgia, terapie locoregionali, terapie sistemiche e controllo delle complicanze.
- L’intervento chirurgico prevede l’esplorazione del pancreas e di tutta la cavità addominale alla ricerca di possibili localizzazioni extrapancreatiche, oltre all’utilizzo dell’ecografia intraoperatoria, metodica utilissima per l’identificazione della neoplasia e la valutazione dei suoi rapporti con le strutture circostanti. L’obiettivo della chirurgia dei tumori endocrini pancreatici è l’asportazione completa del tumore primitivo e delle eventuali metastasi, cercando nello stesso tempo di risparmiare quanto più possibile il pancreas sano. In caso di tumore funzionante si otterrà anche la scomparsa della sindrome clinica. Nel caso sia impossibile un intervento radicale, è utile anche la riduzione di massa, il cosiddetto “debulking” (asportazione della parte di tumore tecnicamente asportabile, lasciando in sede la parte restante che, per la sua aderenza ai grossi vasi addominali, non è possibile asportare).
- La terapia locoregionale comprende diverse procedure, attuabili per via percutanea o durante l’intervento chirurgico: l’embolizzazione (TAE) di rami arteriosi epatici diretti alle metastasi, o la chemioembolizzazione (TACE) che prevede in aggiunta l’infusione locale di farmaci chemioterapici, o l’infusione locale di microsfere di polimeri radioattivi; la distruzione con radiofrequenza (RFA), con iniezione di alcool o mediante microonde (MW).
- La terapia sistemica è necessaria nei pazienti con malattia residua dopo chirurgia non radicale (debulking) e/o terapia locoregionale. Per i tumori funzionanti (ma anche nei tumori non funzionanti dotati di recettori specifici) si usano i derivati a effetto prolungato della somatostatina (analoghi della somatostatina, SS-A). Di recente introduzione sono i farmaci a bersaglio molecolare (come Everolimus e Sunitinib) e la PRRT (radioterapia selettiva per le cellule tumorali endocrine che esprimono recettori per la somatostatina). La chemioterapia è utilizzata per i tumori moderatamente o scarsamente differenziati. Temozolamide e Capecitabina sono farmaci orali usati nei tumori a rapida progressione; mentre Cisplatino ed Etoposide, o 5-Fluorouracile e Streptozocina, sono utilizzati per i tumori scarsamente differenziati. In casi molto selezionati è stato impiegato anche il trapianto di fegato, che è però limitato dalla scarsa disponibilità di organi donati.
- Le più comuni complicanze e gli effetti collaterali del trattamento di questi tumori includono l’iperglicemia, il malassorbimento, il deficit di assorbimento di alcune vitamine, che richiedono il trattamento, rispettivamente, con insulina, enzimi pancreatici, supplemento di vitamine. L'eventuale comparsa di metastasi scheletriche, cerebrali o vertebrali richiede invece il ricorso alla radioterapia.
La strategia diagnostica e terapeutica varia in ogni caso in relazione ai diversi tipi di tumore endocrino del pancreas e richiede grande esperienza di equipe multidisciplinari.
L’insulinoma è la neoplasia endocrina pancreatica più frequente. Il tumore interessa con la stessa frequenza le varie porzioni del pancreas, e nel 10% dei casi fa parte di una sindrome familiare associata ad altre neoplasie endocrine (MEN 1). La presenza di metastasi ai linfonodi peripancreatici o al fegato è riscontrata nel 10% dei pazienti e denuncia la malignità del tumore. Il quadro clinico è sintetizzato nella triade di Whipple caratterizzata da: 1) sintomi dovuti all’ipoglicemia a digiuno (vedi oltre); 2) livelli di glucosio nel sangue < 50 mg/dl; 3) remissione dei sintomi con l’assunzione di glucosio. La produzione autonoma d’insulina da parte dell’insulinoma è responsabile dell’ipoglicemia con sintomi che possono essere distinti in due categorie:
1) sintomi da neuroglicopenia (carenza di zucchero a disposizione del cervello che è il maggiore utilizzatore del glucosio di tutto l’organismo): incapacità di concentrazione, confusione mentale, cambiamento della personalità e del comportamento, fino al sopore ed al coma che compare abitualmente dopo digiuno prolungato.
2) sintomi da eccesso di produzione di ormoni surrenalici (catecolamine) messo in atto dall’organismo nel tentativo di correggere l’ipoglicemia: palpitazioni, tremori, pallore, sudorazione profusa e tachicardia.
Per prevenire la comparsa dei sintomi, i pazienti imparano, spesso spontaneamente, ad assumere alimenti ricchi di carboidrati ogniqualvolta percepiscono l’arrivo dei disturbi. Si abituano inoltre ad aumentare la frequenza dei pasti, con conseguente aumento anche cospicuo del peso corporeo.
Per porre la diagnosi di insulinoma ci si avvale del test del digiuno prolungato (fino a 72 ore) con prelievo di sangue ogni 4-6 ore e, soprattutto, al momento della comparsa dei sintomi. Sul sangue prelevato si esegue il dosaggio contemporaneo della glicemia e dell’insulina (ed eventualmente della proinsulina) plasmatica e la diagnosi si basa sulla dimostrazione, al momento della crisi ipoglicemica, di un eccesso di insulina rispetto alla glicemia del paziente (con rapporto tra insulina/glicemia superiore a 0.3).
Le indagini mirate alla localizzazione del tumore (ecografia, TAC, RMN) vanno intraprese solo dopo la conferma biochimica della diagnosi di insulinoma e due indagini positive concordi sono considerate sufficienti. L’esecuzione delle indagini mirate alla localizzazione del tumore in assenza della diagnosi può dar luogo a reperti falsamente positivi, che possono a loro volta creare inutili problemi al medico ed al paziente.
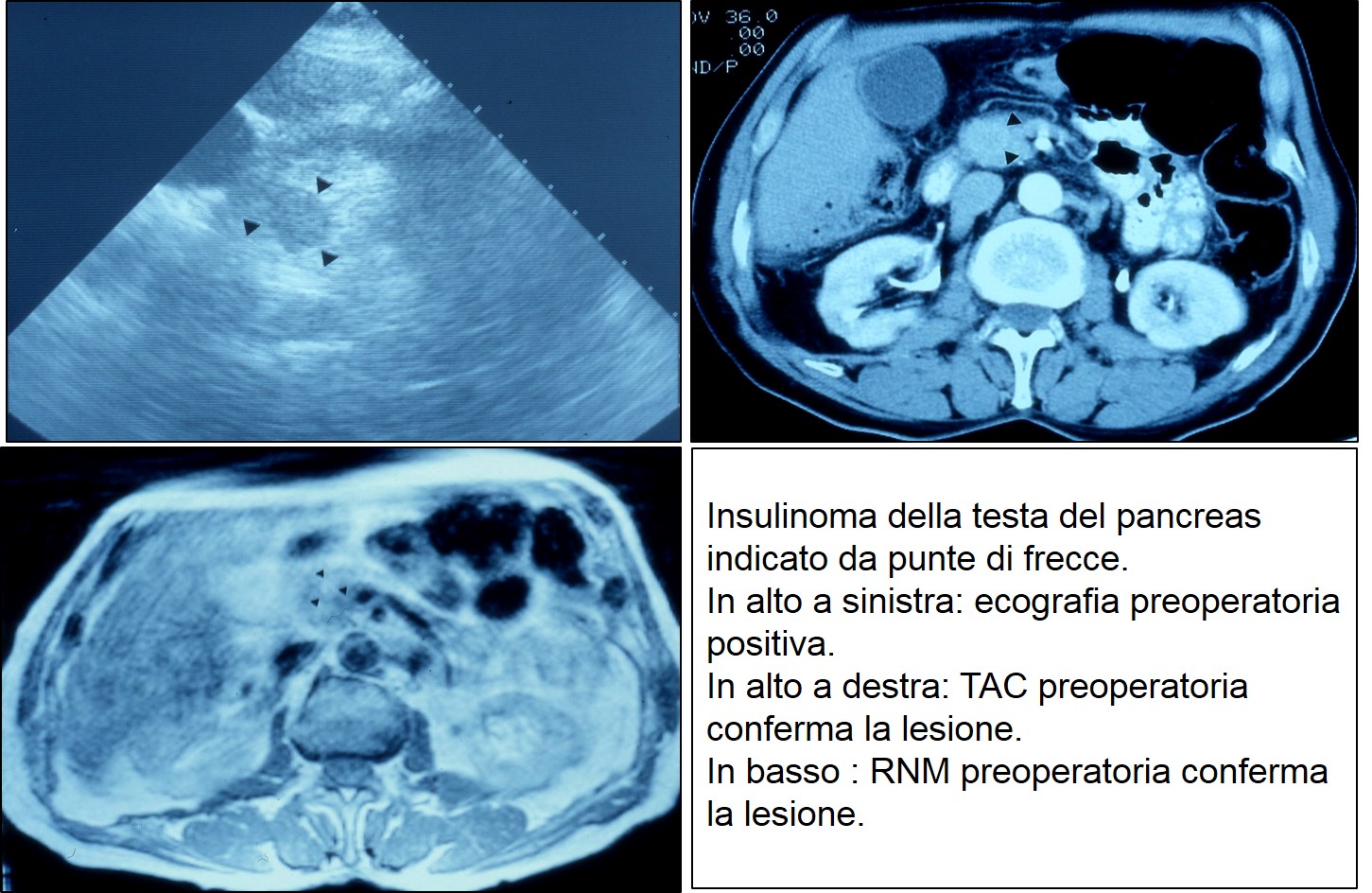
Eco TAC RNM di insulinoma cefalico
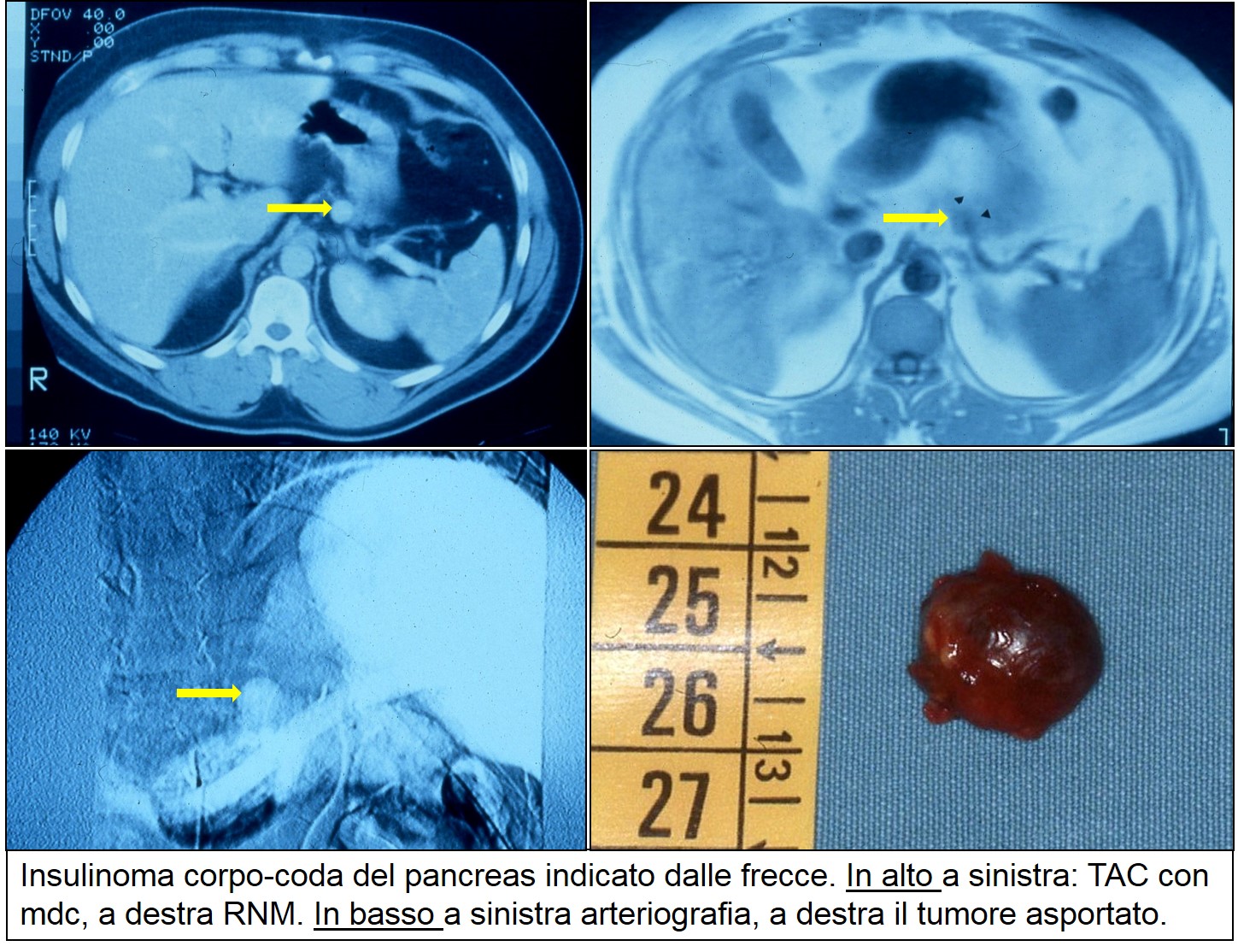
TAC RNM e arteriografia di insulinoma
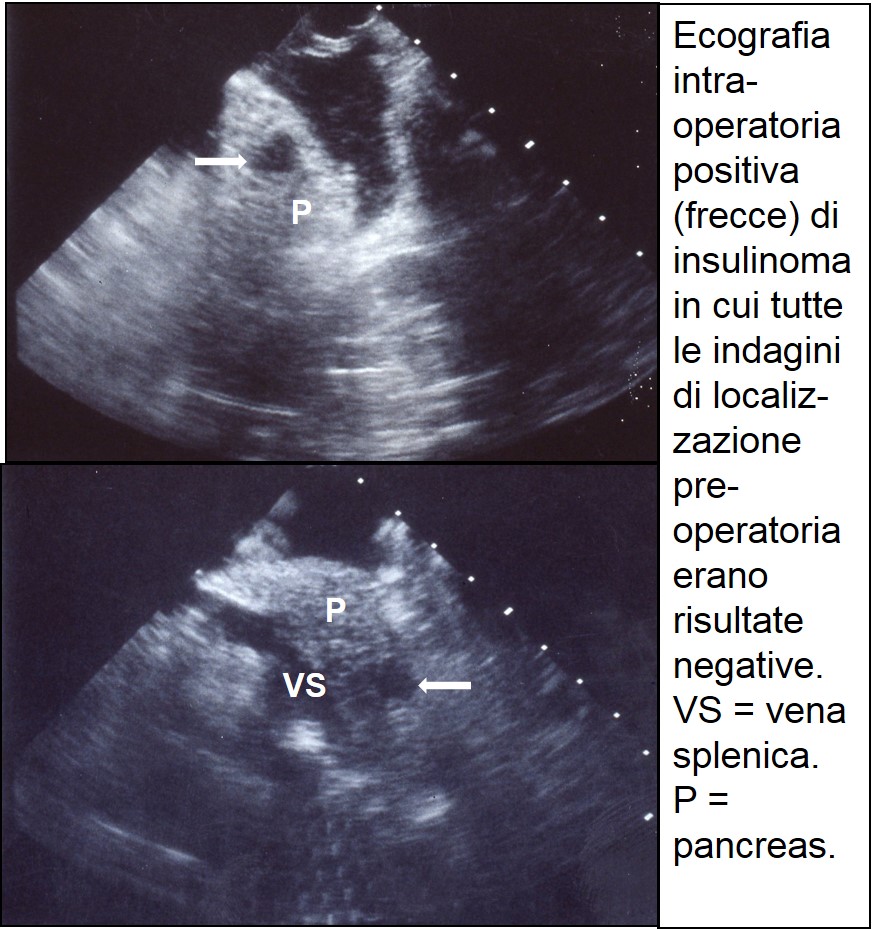
ecografia intraoperatoria di insulinoma
In caso di ripetuta negatività delle indagini, si passa a metodiche diagnostiche più invasive, quali l’ecoendoscopia e/o l’angiografia selettiva con iniezione di calcio (potente stimolatore della secrezione insulinica) nelle tre arterie principali del pancreas (gastroduodenale, mesenterica superiore e splenica). Prelievi seriati del sangue venoso refluo con dosaggio dell’insulina sono in grado di “regionalizzare” la produzione dell’insulina e quindi di indicare la regione pancreatica (testa, istmo, corpo o coda) sede dell’insulinoma. A differenza degli altri tumori neuroendocrini pancreatici, l’insulinoma viene difficilmente identificato alla 68Ga-DOTANOC PET/TAC, data la bassa espressione di recettori ad alta affinità per tale tracciante. Utili per la localizzazione dell’insulinoma risultano invece la 18F-F-DOPA PET/TAC e, di recente introduzione, la scintigrafia (SPECT) o PET/TAC con leganti marcati del recettore glucagonlike peptide1 (GLP1) di cui è ricco l’insulinoma.
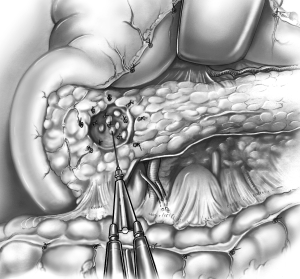
Enucleazione: è evidente la cavità residua all’asportazione del tumore. (dal vol. 6 del trattato di tecnica chirurgica dell’UTET, 2006).
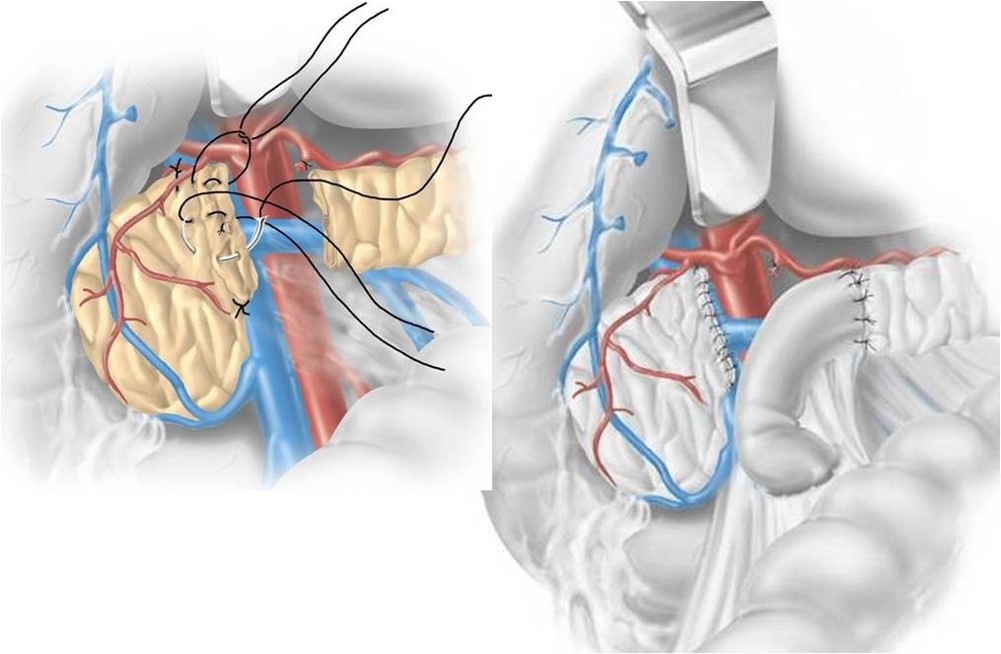
Pancreasectomia intermedia: a sinistra è stato asportato l’istmo del pancreas con il tumore. Il pancreas sul lato destro viene suturato. A destra il corpo-coda del pancreas anastomizzato all’intestino. (dal vol. 6 del trattato di tecnica chirurgica dell’UTET, 2006).
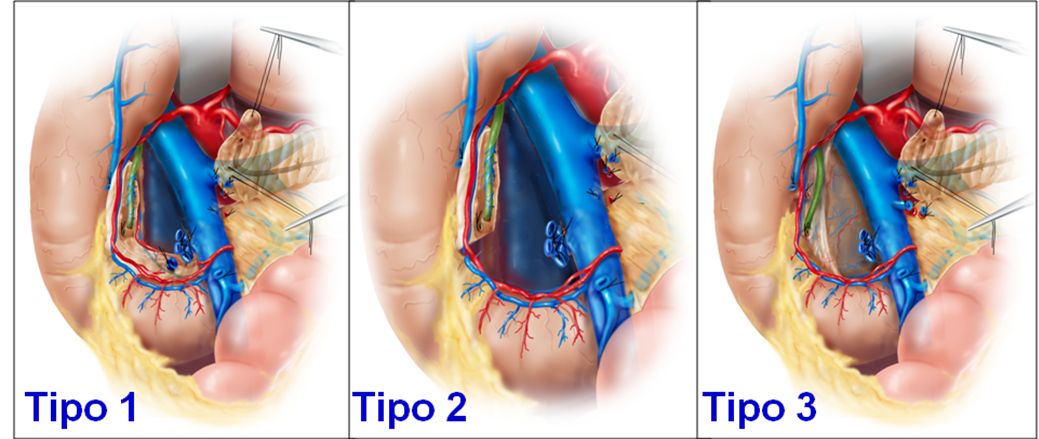
Resezione della testa del pancreas con conservazione del duodeno e del coledoco. Da sinistra a destra: Tipo 1: la testa del pancreas è stata staccata dai vasi mesenterici e dal coledoco (in verde);è stata asportata la testa del pancreas lasciandone alcuni millimetri attaccati al duodeno. Tipo 2: é stato asportato anche il pancreas aderente al duodeno sotto lo sbocco del coledoco, lasciando solo il pancreas tra coledoco e duodeno. Tipo 3: è stato asportato anche il pancreas residuo tra coledoco e duodeno. Il corpo coda del pancreas è anastomizzato all’intestino come per la pancreasectomia intermedia. (dal vol. 6 del trattato di tecnica chirurgica dell’UTET, 2006).
L’asportazione chirurgica del tumore è la terapia di scelta ogni qualvolta possibile, cercando di risparmiare quanto più tessuto ghiandolare sano possibile. Generalmente le piccole dimensioni del tumore e la sua benignità ne consentono la semplice enucleazione, purché il dotto escretore principale (dotto di Wirsung) non sia in prossimità della lesione (per il rischio di danneggiarlo durante la procedura e provocare una fistola pancreatica). In caso contrario l’intervento richiederà una resezione del pancreas più o meno estesa: pancreasectomia intermedia per lesioni centrali, resezione della testa del pancreas con conservazione del duodeno per lesioni della testa del pancreas, resezione della coda con conservazione della milza per lesioni caudali. Per lesioni più voluminose è necessario procedere con resezioni più estese (duodenocefalopancreasectomia, pancreasectomia sinistra).
Il gastrinoma è il secondo più comune tumore endocrino del pancreas (singolo o multiplo, piccolo, localizzato nella testa pancreatica nel 70-90% dei casi) e/o nel duodeno (singolo o più spesso multiplo, sottomucoso, molto piccolo o microscopico). E’ maligno nel 60% dei pazienti e nel 20-25% dei casi è associato a una sindrome familiare di adenomatosi multiendocrina (MEN-1) (vedi più avanti). La maggioranza dei pazienti con sindrome di Zollinger-Ellison (ZES) e MEN-1 ha gastrinomi multipli duodenali e iperparatiroidismo [aumento del paratormone (PTH)]. Nel 10% circa dei pazienti, il tumore è talmente piccolo che non viene trovato nonostante un’accurata esplorazione chirurgica eseguita da una persona esperta. Il quadro clinico è dovuto agli effetti dell’eccesso di secrezione acida basale dello stomaco (malattia ulcerosa gastrointestinale, esofagite e diarrea), legato allo stimolo continuo esercitato dall’ormone gastrina prodotto in modo incontrollato dal tumore. La maggior parte dei pazienti ha una storia di ulcera duodenale da diversi anni, ma nel 8-10% dei casi la diarrea, eventualmente associata a steatorrea (abbondanti sostanze grasse non digerite nelle feci), costituisce l’unica manifestazione. L’avvento dei farmaci anti-H2 [Cimetidina (1976), Ranitidina (1981), Famotidina (1981), Nizatidina (1987) ] e, soprattutto, degli inibitori di pompa protonica [Omeprazolo (1989), Lansoprazolo (1995), Rabeprazolo (1998), Pantoprazolo (2000), Dexlansoprazolo (2009), Esomeprazolo (2010)] ha consentito di dominare agevolmente l’eccessiva produzione di acido da parte dello stomaco. L'efficacia di questi farmaci, in assenza di un metodico dosaggio della gastrina prima di iniziare la terapia, favorisce purtroppo un ritardo nella diagnosi di ZES, dato che essi sono in grado di guarire la maggior parte delle ulcere provocate dal gastrinoma.
La diagnosi di gastrinoma è essenzialmente biochimica. La prima tappa è rappresentata dalla misurazione della gastrina sierica a digiuno e dell’output gastrico acido basale (BAO = Basal Acid Output) interrompendo, se possibile, i farmaci antisecretori. I pazienti con ZES hanno elevati livelli sierici basali di gastrina e ipersecrezione acida gastrica basale (BAO > 15 mmol/h in pazienti non gastroresecati e > 5 mmol/h in pazienti parzialmente gastroresecati). Nel caso in cui la gastrinemia basale e il BAO diano risultati non conclusivi, è utile eseguire i cosiddetti “test di stimolo”. Il principale test di stimolo prevede la somministrazione di secretina (ormone normalmente prodotto dal duodeno) ed è considerato diagnostico di ZES quando, dopo l’iniezione della secretina, si documenta un incremento dei livelli sierici di gastrina di 200 pg/ml o un incremento maggiore del 50% rispetto ai valori basali. Il test con secretina è utile per escludere la ZES in pazienti con gastrinemia elevata (dovuta ad esempio a una gastrite atrofica oppure al trattamento con farmaci antisecretori gastrici), magari in presenza di un BAO elevato dovuto ad altre cause. E’ necessario associare al dosaggio della gastrinemia quello di alcuni marcatori neuroendocrini [Enolasi Neuronale Specifica (NSE), Cromogranina A] e di altri ormoni [Paratormone (PTH), Prolattina (PRL), Ormone della crescita(GH), ecc.] abitualmente aumentati in presenza di una MEN-1.
Una volta posta diagnosi di ZES, si passa alle procedure di localizzazione del tumore. Le tecniche d’immagine comprendono l’ecografia, la TAC, la RMN, l’Ecoendoscopia, la scintigrafia recettoriale con Octreotide (OCTREOSCAN) e, più recentemente, la PET/TAC con 68Ga-DOTANOC.
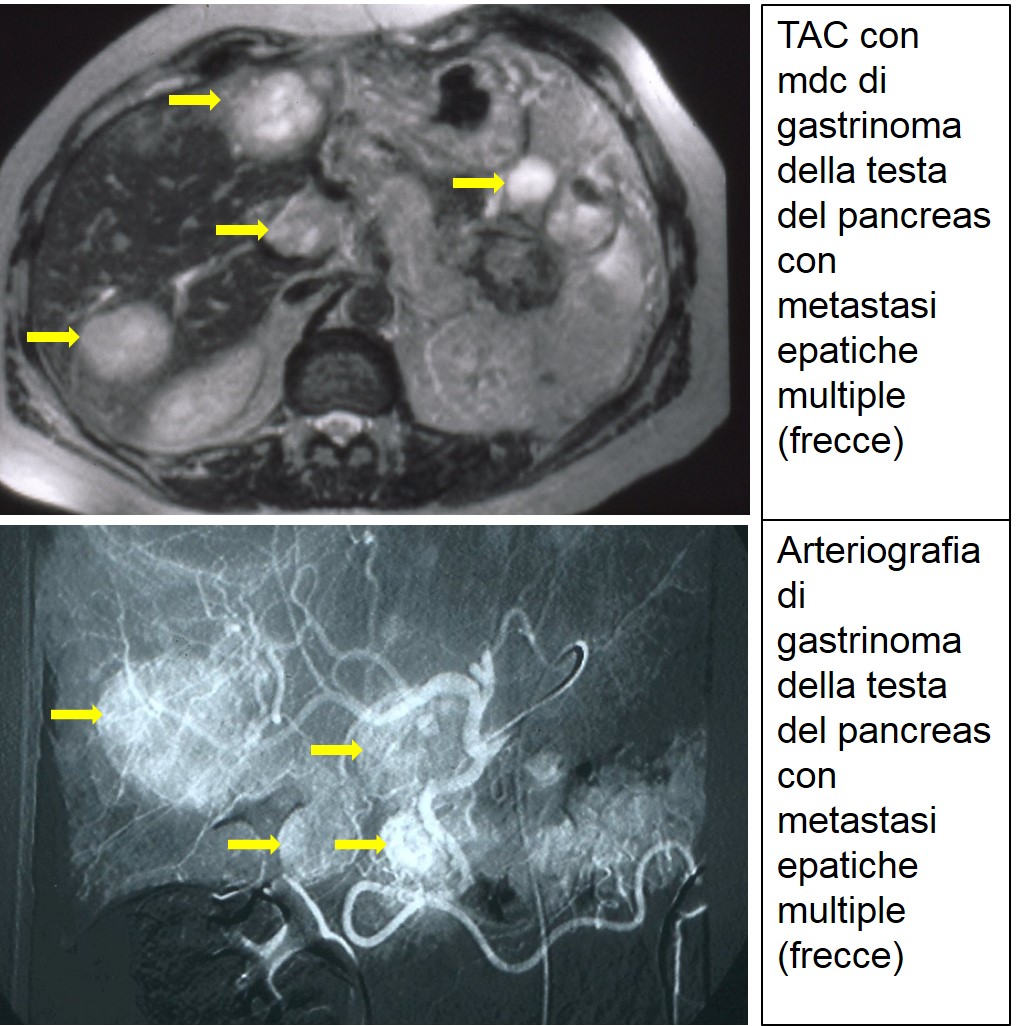
TAC e arteriografia di gastrinoma metastatico
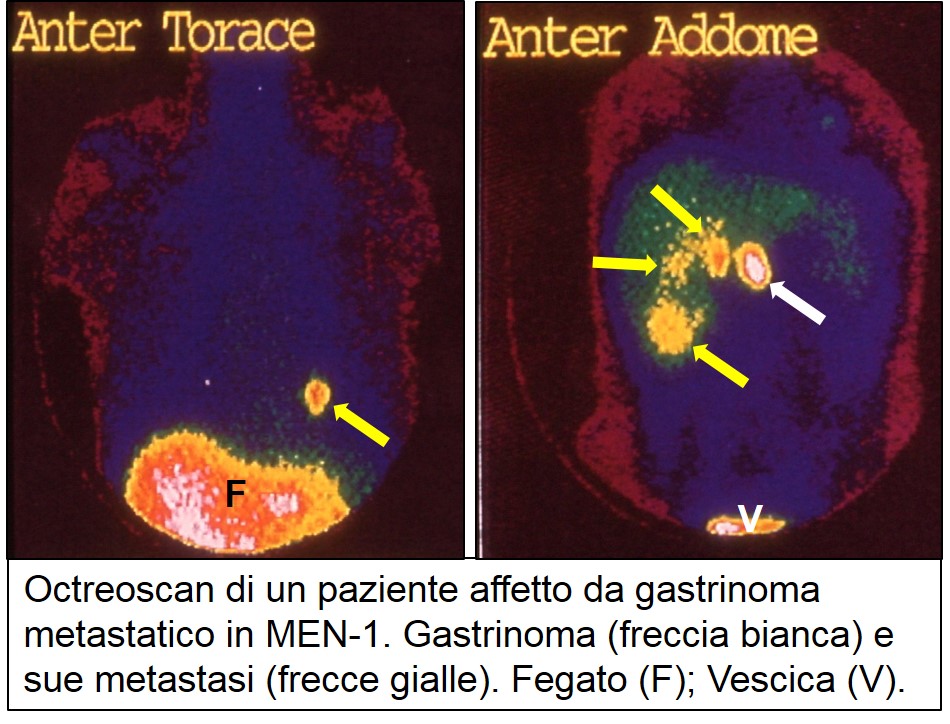
Octreoscan di gastrinoma metastatico
E’ possibile che nessuna delle tecniche elencate riesca a visualizzare il tumore, per cui l’esplorazione chirurgica con metodiche adeguate e grande esperienza va considerata parte delle tecniche di localizzazione.
La terapia medica (con anti-H2 o inibitori di pompa protonica) deve essere iniziata appena possibile e modificata al bisogno fino a ottenere un controllo ottimale della secrezione acida. Prima della scoperta dei farmaci antisecretivi, la diagnosi di ZES imponeva di eseguire il trattamento chirurgico mediante gastrectomia totale (asportazione chirurgica di tutto lo stomaco) appena posta la diagnosi per prevenire il rischio di complicanze mortali della patologia ulcerosa. Attualmente, invece, il trattamento chirurgico è mirato all’asportazione del tumore. La fase iniziale dell’intervento integra le procedure di localizzazione preoperatorie mediante l’esecuzione dell’ecografia intraoperatoria e della duodenotomia con palpazione manuale della parete duodenale. In caso di tumore primario resecabile, il trattamento include l’enucleazione, la resezione duodenale o pancreatica più o meno estesa.

TAC e foto gastrinoma
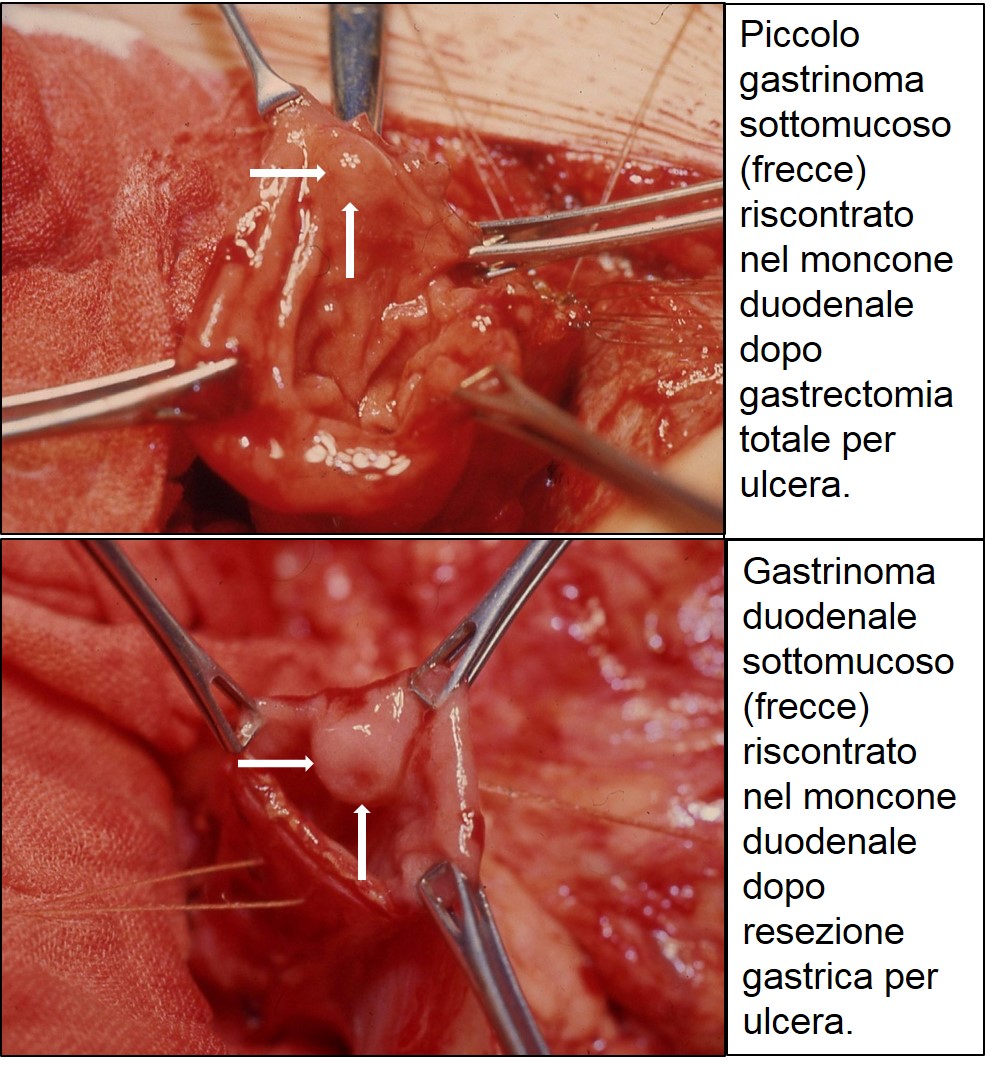
foto di gastrinomi duodenali
In caso di malattia non resecabile in modo radicale, si potranno eseguire interventi di asportazione parziale, mirati a ridurre drasticamente la quantità di tumore residuo. Nei casi di malattia avanzata non suscettibile di intervento chirurgico, oppure nei pazienti sottoposti a intervento di riduzione della massa tumorale, si può ricorrere, a seconda della localizzazione della malattia residua, al trattamento con analoghi della somatostatina, all’embolizzazione delle arterie del fegato dirette alle metastasi epatiche e/o alla chemioterapia, eventualmente associate.
L’associazione fra un tumore pancreatico e una lesione cutanea tipica è stata notata fin dal 1942, ma solo nel 1974 è stato scoperto che il tumore pancreatico, responsabile delle lesioni cutanee, secerne glucagone. Si tratta di un tumore raro (1 caso ogni 20 milioni di abitanti/anno), frequentemente maligno (>80%), voluminoso, localizzato per lo più nel corpo-coda del pancreas. Sono stati descritti anche degli adenomi benigni, abitualmente asintomatici, che costituiscono un reperto occasionale per lo più autoptico. Il quadro clinico è caratterizzato da eritema necrolitico migrante (vedi la figura) (70%), modesto diabete (83%), glossite e stomatite (34%), calo ponderale con appetito conservato (66%), malattia tromboembolica (30%), saltuaria nausea e vomito e disturbi neuropsichiatrici. Gli esami del sangue mostrano anemia normocromica normocitica (85%) e ipoaminoacidemia (100%). La diagnosi preoperatoria si basa sul riscontro di una glucagonemia >1000 pg/ml (valori normali <100 pg/ml), associata a un tumore endocrino del pancreas. In assenza della sindrome tipica la diagnosi è spesso posta sull’esame istologico del pezzo operatorio.
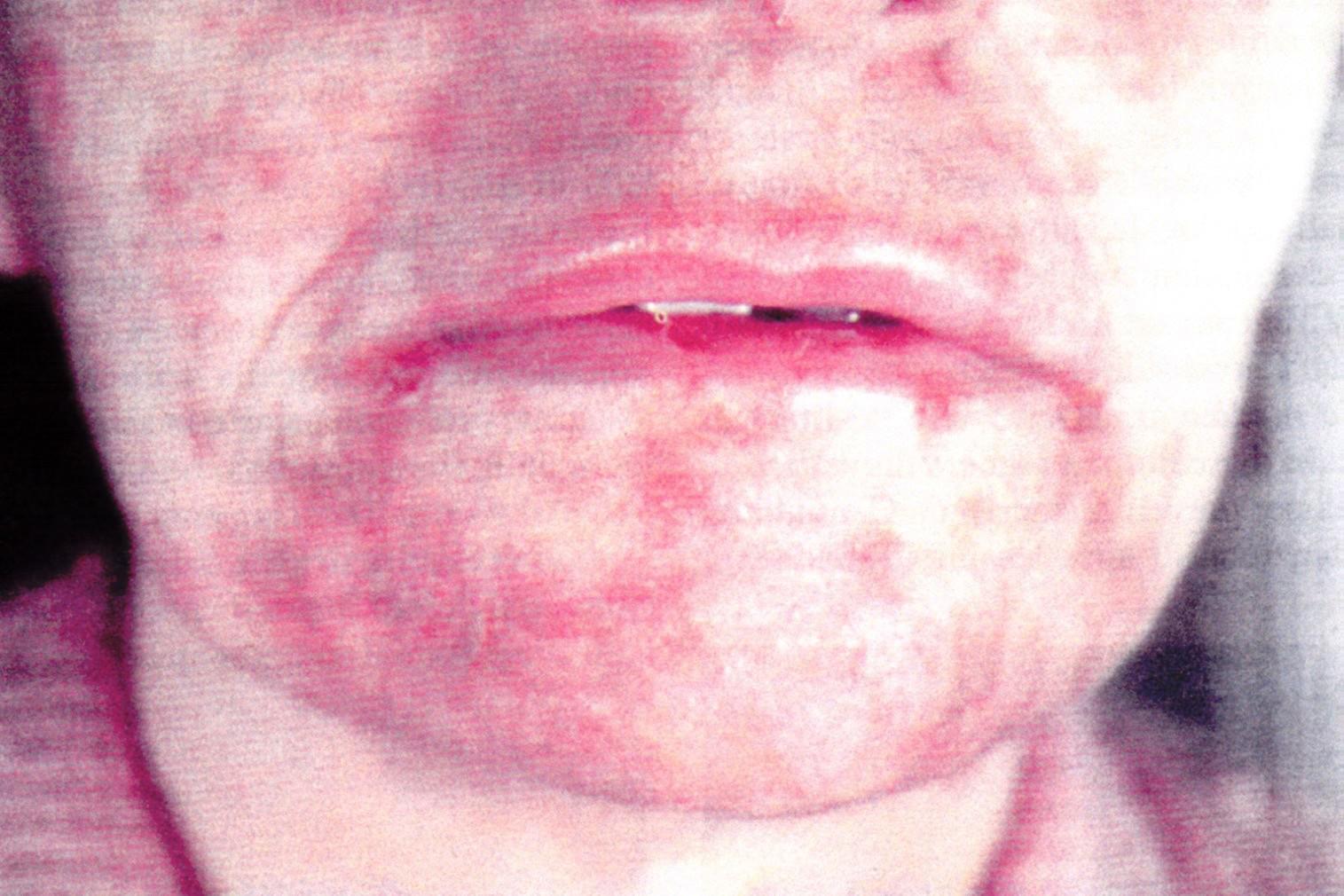
Paziente affetta da Glucagonoma del corpo-coda del pancreas: eritema necrolitico migrante in regione periorale.
Per gentile concessione del Prof. GC Biliotti, Dipartimento di Fisiopatologia Clinica, Firenze.
La localizzazione del tumore, spesso voluminoso e metastatico, è agevolmente ottenuta mediante l’ecografia, la TAC e/o la RMN, oppure tramite ecoendoscopia, in caso di tumori molto piccoli.
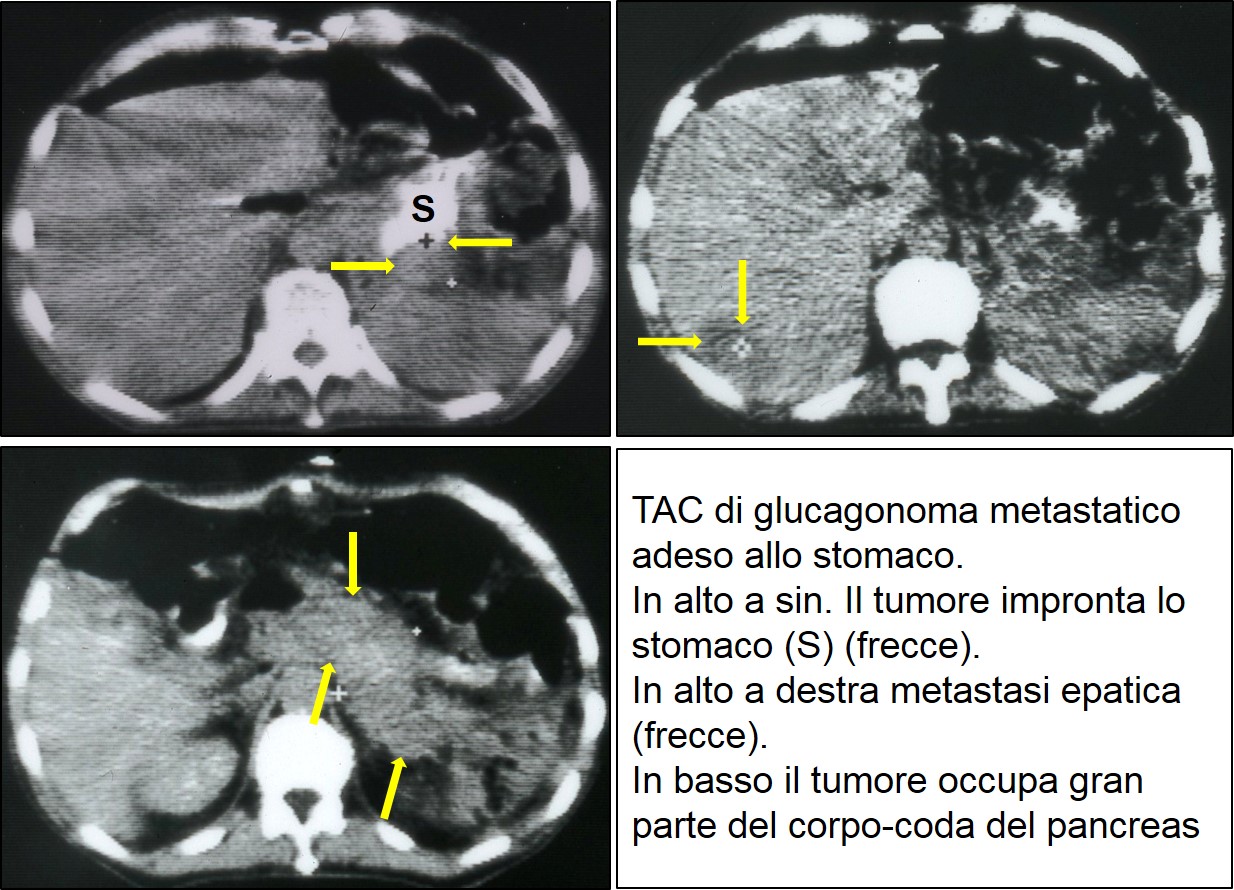
TAC di glucagonoma metastatico
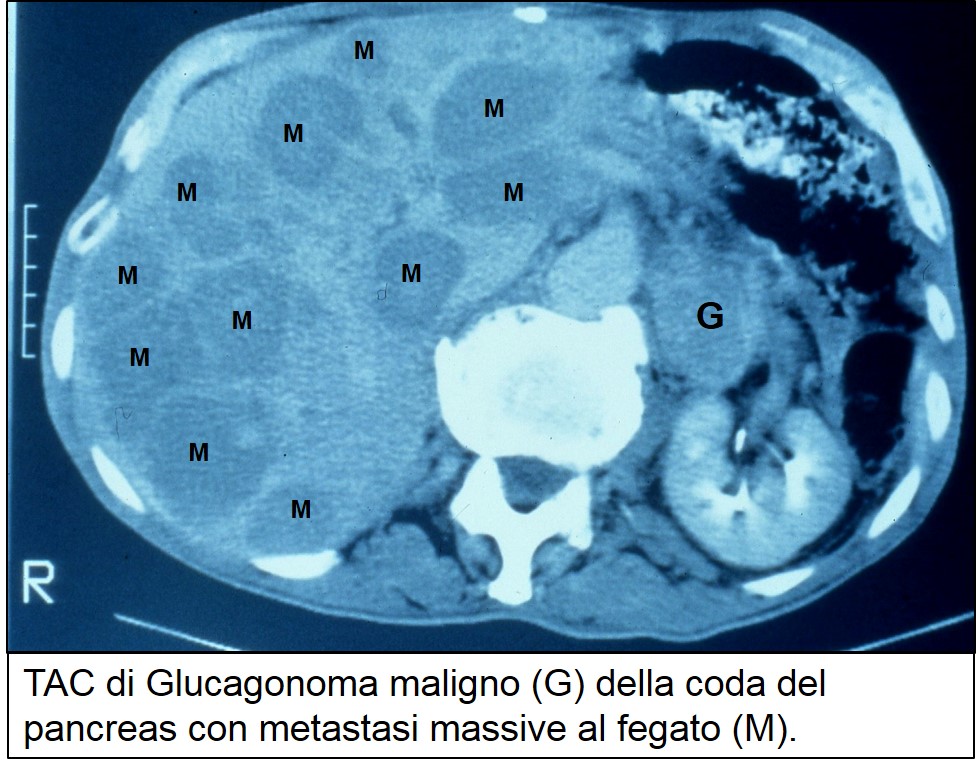
TAC di glucagonoma con metastasi massive
Si usano anche metodiche di imaging funzionale per la diagnosi di sede e per la stadiazione (scintigrafia con Octreoscan o 68Ga-DOTANOC PET/TAC). La terapia di scelta è chirurgica ed è risolutiva solo nel 30% dei casi;
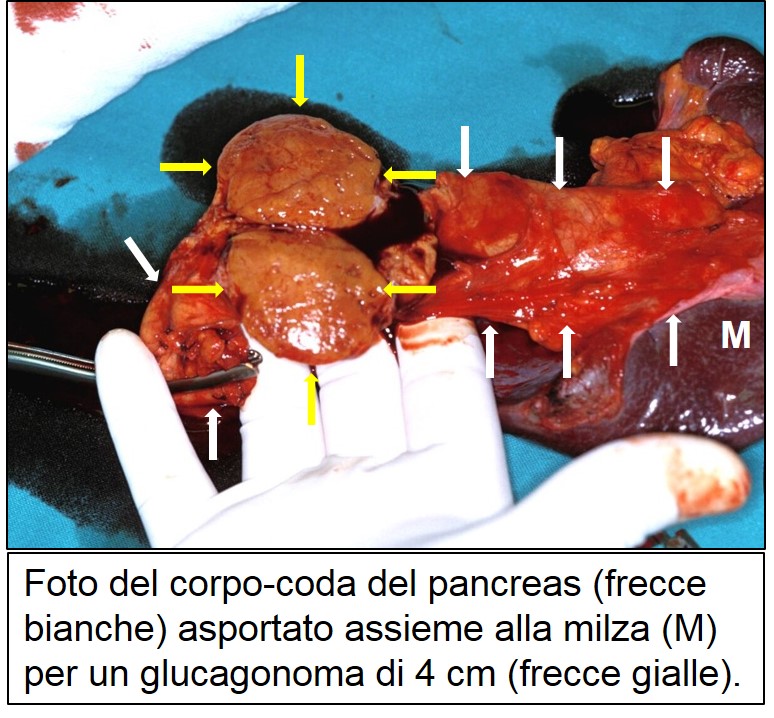
foto pezzo opera di glucagonoma 4 cm
anche un intervento demolitivo non radicale (debulking) è utile per migliorare la qualità di vita e prolungare la sopravvivenza del paziente. La terapia medica con analoghi della somatostatina è efficace sulla sindrome clinica nella maggior parte dei pazienti. La chemioterapia con streptozotocina, 5-Fluorouracile e doxorubicina offre occasionali risposte. Più recentemente, l’uso della dacarbazina (DTIC) ha permesso di ottenere risposte obiettive notevoli e prolungate che, in alcuni casi, hanno reso possibile una successiva exeresi chirurgica apparentemente radicale in pazienti in precedenza inoperabili; essa è ora considerata il farmaco di scelta.
Si tratta di una sindrome rara (1 caso ogni 10 milioni di abitanti/anno), attribuita all’eccessiva secrezione dell’ormone VIP (Polipeptide Intestinale Vasoattivo) da parte di un tumore pancreatico (80% dei pazienti) o di una microadenomatosi o carcinomatosi insulare (10% dei casi) o, soprattutto in età pediatrica, di un tumore localizzato al di fuori dell’apparato digerente (ganglioneuroma surrenalico, retroperitoneale o del collo, 10% dei casi). Il quadro clinico è caratterizzato da diarrea acquosa, accompagnata da ipokaliemia e acloridria. Si può sospettare la sindrome solo quando il volume delle feci supera i 700 ml/die e la diarrea persiste anche a digiuno, con grave perdita di potassio e bicarbonati. La sindrome clinica non ha un andamento continuo, ma si manifesta con episodi più o meno prolungati, alternati a periodi di benessere di durata variabile. La diagnosi si basa sulle caratteristiche della diarrea, sulla dimostrazione di elevati livelli di VIP (>200 pg/ml, con valori normali inferiori a 170 pg/ml) e sul riscontro di un tumore possibile produttore di VIP. La localizzazione del tumore mediante ecografia o TAC è di solito agevole, poiché si tratta in genere di tumori voluminosi, mentre l’ecoendoscopia è utile nel caso di tumori più piccoli. Si usano anche metodiche di imaging funzionale per la diagnosi di sede e per la stadiazione (scintigrafia con Octreoscan o 68Ga-DOTANOC PET/TAC). La terapia è inizialmente medica e si avvale di idratazione parenterale e analoghi della somatostatina, per arrestare la diarrea e riequilibrare il paziente che, a causa dei bassi livelli di potassio nel sangue, corre notevoli rischi cardiologici, specie se sottoposto a intervento chirurgico. Si procede quindi all’intervento chirurgico, che, secondo l’estensione della malattia, può essere radicale (con conseguente controllo ottimale della sindrome clinica), oppure può limitarsi alla riduzione della massa tumorale (debulking) nei pazienti in cui l’exeresi radicale non è praticabile. La terapia chirurgica va integrata con la chemioterapia; l’associazione di Streptozotocina e 5-Fluorouracile ottiene una risposta positiva nel 60-70% dei casi, risposta che, purtroppo, è di breve durata.
Il somatostatinoma è un tumore raro e uno dei più recenti tumori endocrini identificati. A sede pancreatica nel 40% dei casi (e duodenale nel restante 60%), maligno nell’80-85% dei casi, può comparire associato alla sindrome di von Recklinghausen (neurofibromatosi) nel 40% dei somatostatinomi duodenali. La somatostatina inibisce sia l’immissione in circolo sia l’attività della maggior parte degli ormoni gastrointestinali sui rispettivi organi bersaglio. Il quadro clinico può essere dovuto alla secrezione ormonale (sindrome da somatostatinoma) e/o alla compressione della massa tumorale sugli organi circostanti. La sindrome è caratterizzata da modesto diabete, diarrea con steatorrea (abbondanti sostanze grasse non digerite nelle feci), ipocloridria, digestione difficile, senso di stomaco pieno dopo i pasti, calcolosi della colecisti, calo ponderale e, occasionalmente, anemia. La compressione della massa tumorale sugli organi circostanti è abitualmente la principale responsabile dei sintomi lamentati dal paziente: ittero per ostruzione delle vie biliari, vomito per ostruzione duodenale e calo di peso. La diagnosi si avvale del dosaggio della somatostatina nel plasma (valori normali < 100 pg/ml) soprattutto per quanto riguarda il tumore pancreatico, mentre è di rara utilità per quello duodenale. Trattandosi abitualmente di tumori voluminosi, la localizzazione del tumore è agevolmente eseguita con l’ecografia e la TAC e/o la RMN, ma per le lesioni più piccole è utile l’ecoendoscopia.
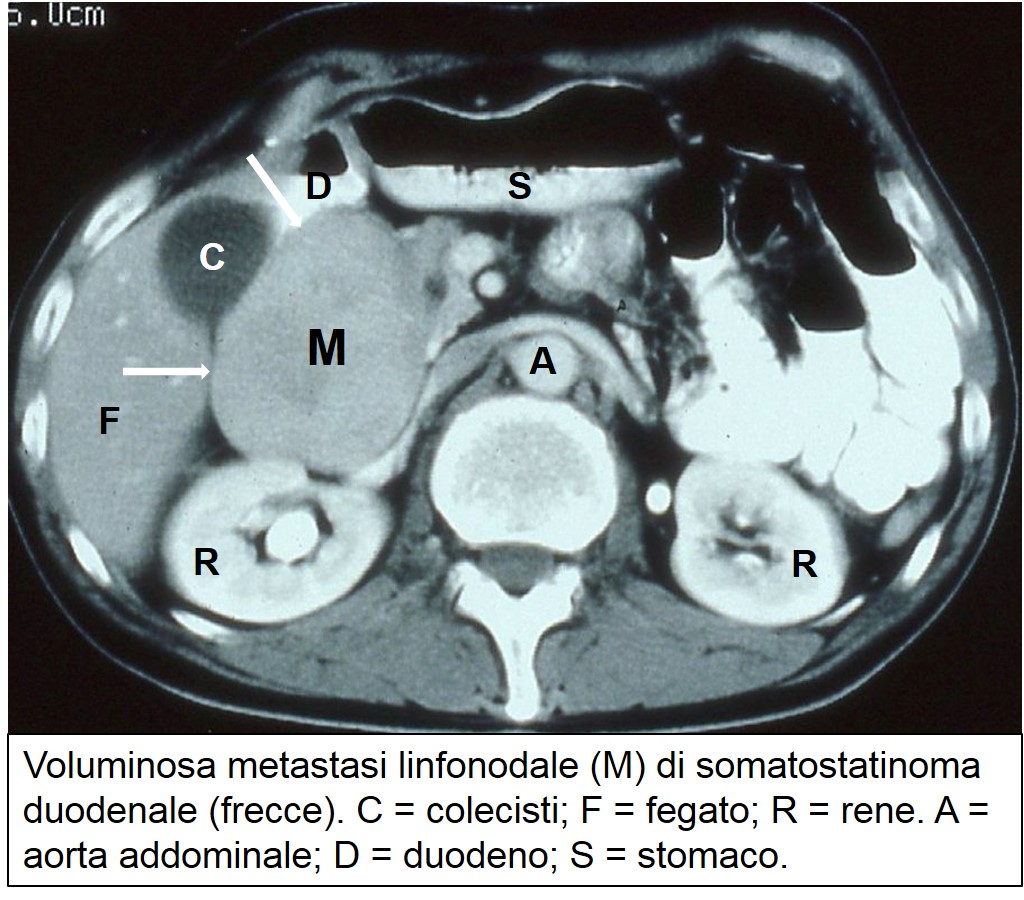
meta linfonodale di somatostatinoma
Si usano anche metodiche di imaging funzionale per la diagnosi di sede e per la stadiazione (scintigrafia con Octreoscan o 68Ga-DOTANOC PET/TAC). La terapia di scelta è chirurgica e mira alla rimozione completa del tumore. Anche l’asportazione parziale, mirata alla riduzione di massa (debulking), è giustificata, poiché migliora la sopravvivenza e la qualità di vita del paziente. In presenza di metastasi non asportabili o di tumore inoperabile o di residuo di malattia dopo riduzione di massa (debulking), può essere presa in considerazione la chemioterapia con i farmaci usati abitualmente per le neoplasie endocrine pancreatiche maligne (vedi parte generale).
Circa tre quarti dei tumori endocrini pancreatici attualmente diagnosticati non è associato a una sindrome clinica. Si tratta di un cambiamento notevole rispetto a 30-40 anni fa, quando rappresentavano solo un quinto dei tumori endocrini allora diagnosticati. Un così rilevante cambiamento è legato sia a una migliore conoscenza del tipo di patologia sia a strumenti diagnostici più sofisticati (vedi la parte generale sull’uso dell’ecografia, della TAC e della RMN nel capitolo sul cancro del pancreas) che hanno consentito di diagnosticare molti tumori endocrini non secernenti di piccole dimensioni e clinicamente asintomatici che precedentemente sfuggivano alla diagnosi. . Il quadro clinico dipende dalla sede e dal volume del tumore e può essere caratterizzato dalla compressione sul coledoco (con ittero), sul duodeno (con nausea e vomito), e dalla eventuale presenza di dolore e calo ponderale. Esiste anche una percentuale di tumori endocrini di piccole dimensioni (<2 cm) clinicamente silenti e scoperti nel corso di esami per altre cause (vedi più avanti). Al momento della diagnosi i tumori endocrini non funzionanti presentano in genere grandi dimensioni e, spesso, metastasi epatiche. In questi casi il quadro clinico è sovrapponibile a quello del cancro del pancreas esocrino, rispetto al quale deve essere condotta un’accurata diagnosi differenziale. A questo scopo è indispensabile documentare istologicamente tutti i tumori maligni del pancreas, specialmente se il decorso della malattia, e le condizioni generali del paziente, non corrispondono a quelle attese, a parità di stadio, per il cancro del pancreas. Rispetto ai tumori esocrini del pancreas, essi hanno in genere una crescita più lenta (40% di sopravvivenza a 5 anni) e la possibilità di essere resecati nel 20% dei casi. Va tuttavia sottolineato che, grazie all’affinarsi e alla diffusione delle tecniche d’immagine (ecografia, TAC, RMN), circa la metà dei tumori endocrini non funzionanti è scoperta casualmente a seguito d’indagini eseguiti per altre patologie. La localizzazione e la stadiazione sono agevoli grazie all’ecografia, alla TAC e alla RNM.
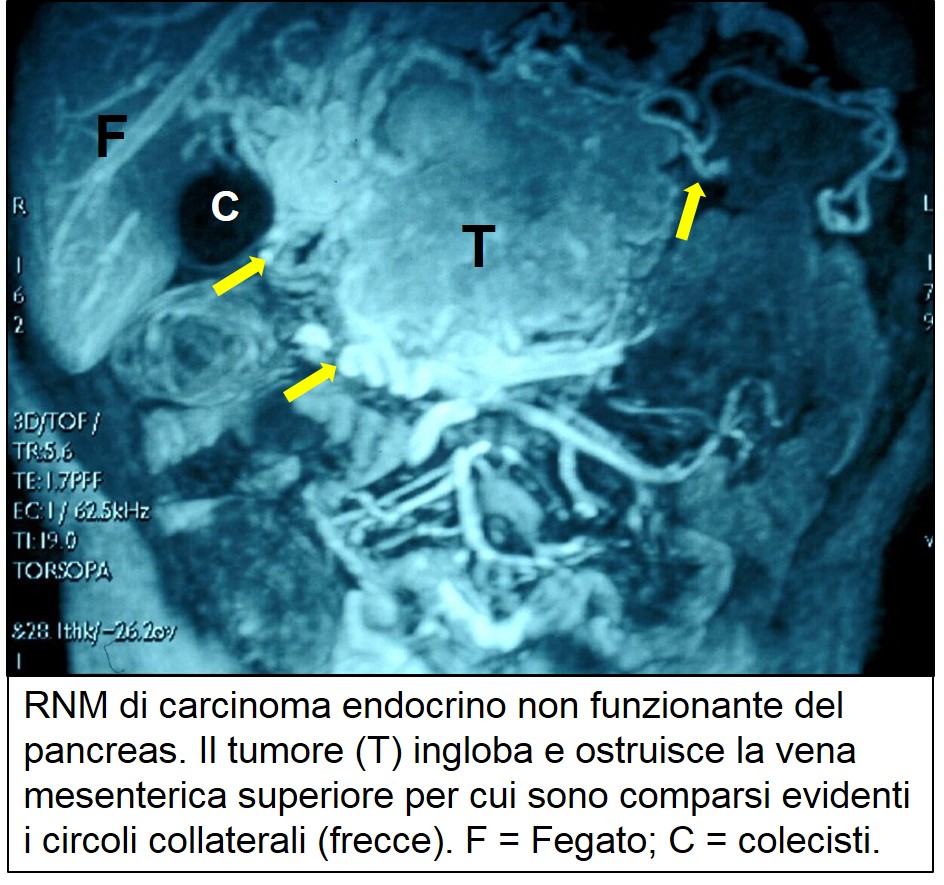
TAC di TEP non funzionante localmente avanzato
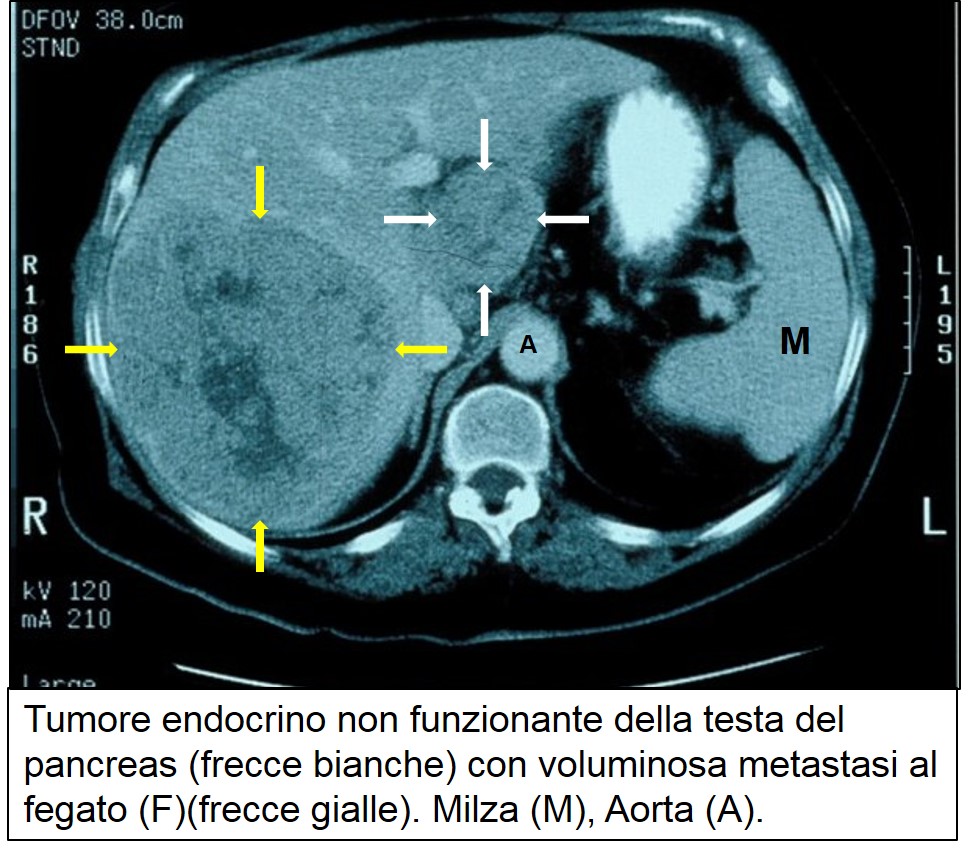
TAC di TEP non funzionante metastatico
E’ importante eseguire preoperatoriamente una scintigrafia con Octreoscan, o meglio una 68Ga-DOTANOC PET/TAC, sia per la successiva scelta terapeutica (presenza/assenza di recettori per la somatostatina) sia per l’identificazione di eventuali recidive in corso di follow up (controlli periodici a distanza).
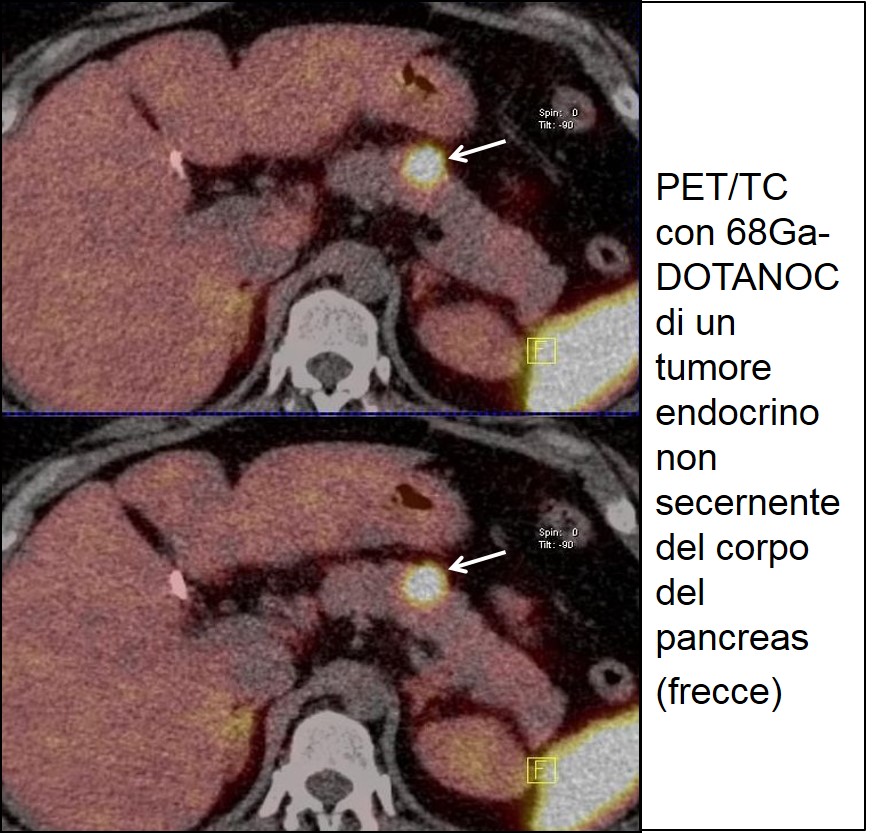
68Ga-DOTANOC di TEP non funzionante
La terapia di scelta nei tumori sintomatici, e di quelli di diametro superiore a 2 cm, è chirurgica. Nelle forme ben differenziate è utile una maggiore aggressività nel trattamento chirurgico (rispetto al carcinoma pancreatico) e anche un intervento di riduzione di massa (debulking) può prolungare la sopravvivenza. La risposta alla chemioterapia con gli abituali farmaci per le neoplasie endocrine pancreatiche maligne (Streptozotocina, 5-Fluorouracile, Interferone) è piuttosto modesta. E’ stata riportata una buona percentuale di risposta in tumori endocrini pancreatici maligni privi di recettori per la somatostatina (68Ga-DOTANOC-PET/TAC negativa) al trattamento con Etoposide e Cisplatino.
Un discorso a parte richiedono i tumori endocrini di piccole dimensioni (<2 cm) scoperti casualmente in corso d’indagini eseguite per altre patologie. Dato che la maggior parte di questi è benigna e non ci sono informazioni adeguate sulla loro evoluzione, è preferibile evitare il trattamento chirurgico, specie se il rischio chirurgico è aumentato per le condizioni generali del paziente e/o l’aspettativa di vita è limitata. In questo caso il paziente viene seguito con esami morfologici periodici (Eco, TAC; o RMN) per rilevare eventuali cambiamenti del volume del tumore. Nei casi dubbi è utile eseguire una 18F-FDG PET/TAC, che consente di identificare i tumori a elevata attività metabolica, probabilmente più aggressivi nonostante le ridotte dimensioni, e proporne l’asportazione.
La sindrome MEN-1 è una rara malattia genetica autosomica dominante causata dall’inattivazione del gene oncosoppressore MEN-1, con conseguente alterazione nella produzione di una proteina chiamata menina. Non esiste una correlazione fissa tra genotipo (DNA) e fenotipo (quadro clinico), tanto che parenti della stessa famiglia con lo stesso difetto genetico hanno manifestazioni cliniche diverse. Nei pazienti con MEN-1 troviamo in varia sequenza l’iperparatiroidismo (dovuto per lo più a iperplasia, raramente a un adenoma, delle paratiroidi), tumori ipofisari (Prolattinoma, ACTHoma, etc.) e tumori endocrini del pancreas (gastrinoma, insulinoma, glucagonoma, etc.). Solo il 50% dei pazienti sviluppa dei tumori endocrini pancreatici macroscopicamente rilevabili, ma tutti presentano iperplasia, displasia e microtumori delle cellule endocrine. I tumori pancreatici di più frequente riscontro sono i gastrinomi, gli insulinomi e i tumori non funzionanti. Il quadro clinico è legato al tipo di tumore presente (funzionante o non funzionante). In assenza di familiarità, il sospetto clinico di MEN-1 si pone in presenza dell’interessamento di almeno due delle tre sedi (pancreas, paratiroidi, ipofisi) tipiche della sindrome. In un paziente con familiarità per MEN-1 la comparsa di una delle lesioni indica che il paziente è portatore della mutazione. Non tutti i familiari di un paziente MEN-1 sono affetti dalla sindrome. L’esame del DNA consente di dirimere i soggetti sani da quelli in cui l’esame genetico sia risultato positivo per la presenza della mutazione del gene MEN-1. Questi ultimi devono essere classificati come MEN-1 indipendentemente dalla presenza di una lesione tipica. La maggior parte dei tumori endocrini pancreatici in MEN-1 sono benigni, ma quando superano i 20 mm di diametro possono comparire metastasi. La localizzazione dei tumori endocrini nella MEN-1 prevede l’uso delle tecniche d’immagine abitualmente utilizzate per i tumori endocrini del pancreas (ecografia, TAC, RMN, ecoendoscopia).
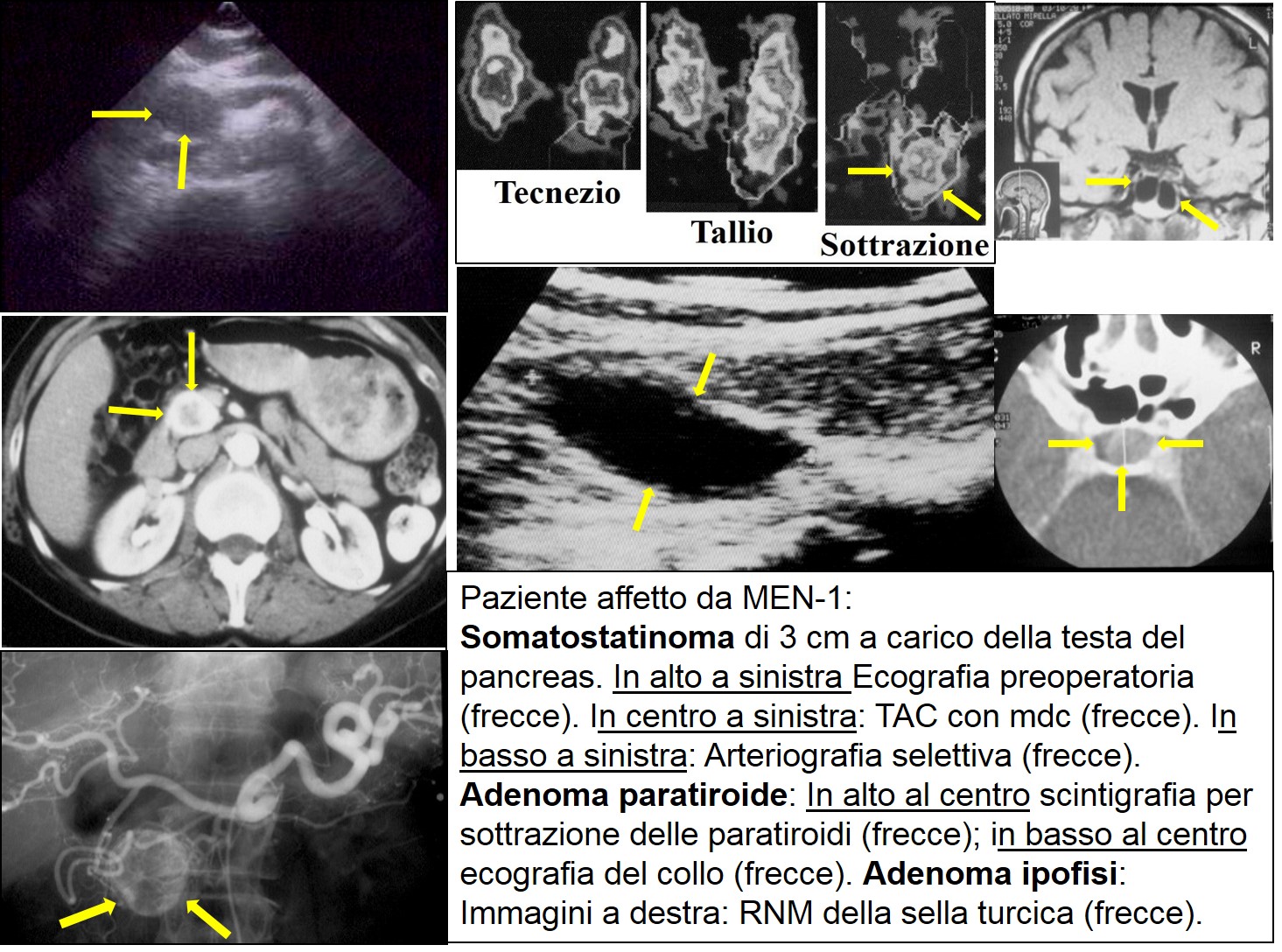
MEN-1 Pancreas Paratiroide Ipofisi
In caso d'intervento, è importante eseguire preoperatoriamente una scintigrafia con Octreoscan, o meglio una 68Ga-DOTANOC PET/TAC, sia per la successiva scelta terapeutica (presenza/assenza di recettori per la somatostatina) sia per l’identificazione di eventuali recidive in corso di follow up. La terapia è chirurgica per i tumori funzionanti (insulinomi, gastrinomi) e per i tumori non funzionanti pari o superiori a 20 mm; invece, i tumori non funzionanti inferiori a 20 mm possono essere messi in follow-up. Poiché esiste una predisposizione genetica alla comparsa di questi tumori, i pazienti devono essere sottoposti a regolare follow-up per scoprire tempestivamente eventuali nuovi tumori e gli interventi chirurgici attuati devono mirare al massimo risparmio di tessuto ghiandolare sano, data la possibilità di eseguire interventi chirurgici ripetuti nel tempo.
La sindrome di von Hippel-Lindau (VHL) è una rara malattia genetica autosomica dominante caratterizzata dallo sviluppo di multiple cisti e di tumori benigni e maligni. Questi tumori sono localizzati nel sistema nervoso centrale e nella retina (emangioblastoma), nel surrene (feocromocitoma), nel rene (carcinoma) e nel pancreas (tumori endocrini e cistici). La malattia è determinata dall’inattivazione del gene oncosoppressore VHL, con conseguente aumentata crescita dei diversi tipi di tumore favorita dallo sviluppo di microvasi sanguigni. Le lesioni pancreatiche tipiche sono cisti singole o multiple e cistoadenomi sierosi (35%-75% dei casi), per la maggior parte benigne. L’incidenza di tumori endocrini (in genere non funzionanti) nella VHL si aggira intorno al 17%, con metastasi presenti solo nel 11-20% dei casi, grazie alla diagnosi precoce attuata mediante screening per la sindrome VHL. La diagnosi si basa sulla scoperta di uno dei tumori tipici della malattia (emangioblastoma retinico, emangioblastoma cerebrale, feocromocitoma, carcinoma renale, tumore endocrino e/o cistoadenoma sieroso del pancreas), specie se in presenza di familiarità per VHL. La localizzazione e stadiazione del tumore pancreatico si attua mediante ecografia, TAC e RMN. E’ importante eseguire preoperatoriamente una scintigrafia con Octreoscan, o meglio una 68Ga-DOTANOC PET/TAC, sia per la successiva scelta terapeutica (presenza/assenza di recettori per la somatostatina) sia per identificare, se positiva, eventuali recidive in corso di follow up. La terapia, dato il potenziale maligno di questi tumori, è chirurgica per tumori superiori a 1 cm.
La rarissima malattia di Mahvash, provocata dall’inattivazione omozigote (e quindi completa) del gene per il recettore del glucagone, è stata scoperta recentemente. Si tratta di una malattia ereditaria autosomica recessiva, caratterizzata da iperplasia delle cellule alfa (che producono glucagone) delle isole di Langerhans, comparsa di tumori endocrini pancreatici e livelli di glucagone estremamente elevati, in assenza della sindrome da glucagonoma (poiché manca il recettore per rendere efficaci gli elevati livelli di glucagone). La diagnosi di malattia è pertanto casuale e legata al riscontro occasionale di uno o più tumori endocrini pancreatici in corso d’indagini morfologiche (ecografia, TAC, RMN) eseguite per motivi non inerenti al pancreas, in associazione a livelli molto elevati di glucagone. La terapia per il momento è chirurgica e mirata all’asportazione dei tumori endocrini. E’ importante un accurato follow-up del paziente dato l’elevato rischio di comparsa di nuovi tumori endocrini pancreatici.